

�߉��ЙC�Ǽ����ڸ������c����x��늳�
�c�o�C������ȣ��ЙC���ϸ��A���γɷ��Ӿ��w�������ڽY���ϸ��Թ̺�ͨ�ã������׃�����Ը��͵������ɱ��M�У��������_�ŵ��ЙC���w�����Ҳ���ܾ��и��ߵ���x���w���ʡ��������ڲ�ͬ����x�Ӵ惦�r����Li+��Na+��K+�����ЙC����ͨ�����F�����ߵķ����ԡ�Ȼ�����M�ܾ���늻��W���Ե��c/��x���մɣ��o�C���������S�����������ֹ���]���c�x���ЙC���O���ϵ��_�l�����������ֹ߀�]�кϳɺ͜yԇ��x���ЙC�ࡣ����������x�Ӄ��Ӳ��ϣ�cation-reservoir material��֮�⣬�ЙC��x�Ӄ��ӻ��W������cation-reservoir chemistries���ă��ڃ������������и��ߵ�����߀ԭ�λ���Ķ���ζ�������ܶȵ����ӡ�n���ЙC�c�x�ӻ�������������߀ԭ�λ�ӽ�2.7 V vs Na+/Na�����ЙCK�x�����O��ֵ�^�͡���ˣ��о��܉��ڸ߉����F�����c/��x�Ӵ惦�����ͺ��������O�ѳɞ����е������⣬ֵ��ע����ǣ��mȻ��x�����|��Li+�cNa+�cK+����늺ɴ惦���ܺ��������W���|��Ӱ��ǿ����A�ϵģ���������ͬ������O�����c���õ��о����١�
�ڴˣ����ߌ��_�l��������x�ӻ��������O�Uչ����Na�ͺ�K���ЙC�ࡣ��-1,2,4,5-�Ļ��ļ�����������x�ӣ�PTtSA4-�����x�鹤���ЙC�Ǽܣ��״β��ú��A������x�ӵ��ЙC늘O�ࣨLi4-PTtSA, Na4-PTtSA��K4-PTtSA)�M���˽�B��ϵ�y�о����l�F�A������x�ӣ�Li+��Na+��K+)�ڷ��Ӻ���ˮƽ���@��Ӱ�늺ɂ������Y�����Լ��������W��늻��W���|���@Щϵ�y�о��Y��������_�l�߉A�x�Ӵ惦���ܵĺ��m�ЙC���O�����ṩ����Ҫ������
�����ݱ�����
1. ���ϵĽY��
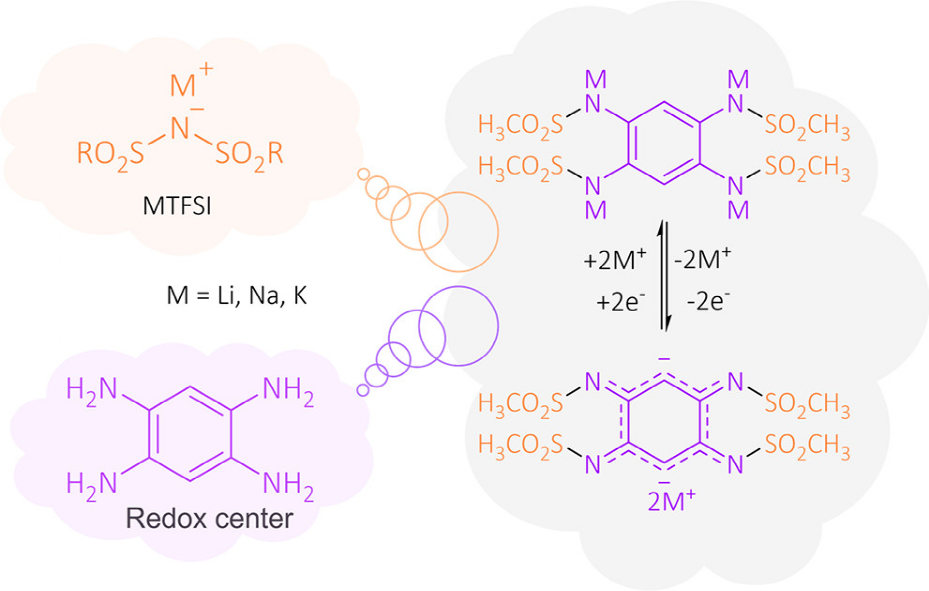
�D 1. �OӋ�ͺϳ�M4-PTtSA�A����x�Ӄ�����ĺ����M�ɡ��Y��������߀ԭ�C�ơ�
�ڈD1�У��ЙC����x�ӹǼܵ��OӋ�`�Ё��ԏV��ʹ�õ�MTFSI��M = Li��Na��K���}��Na4/K4-PTtSA������ͨ�^��ǰ�����Li4-PTtSA�ĸ��M�����ϳɡ�ͬ�r���������ȣ�Ҳ�Ƃ�ͷ�����Li4-PTtSA��
�D2a�@ʾ���S�����w��x�ӵ��x�Ӱ돽����Li+��Na+��K+�������ӣ��Y���ȵõ����ơ����r�x�Ӱ돽���O�����ʺ���λ�������Á�����@Щ�����������Ҫ�Mһ���о��������˽������P�ԡ��ڟo�C���O����Ҳ�^�쵽��Ƶ�څ�ݣ���Li2MoS4��Na2MoS4�� K2MoS4��ǰ���ǟo���εģ��������ǽY���ģ���
2. ���ϵı���
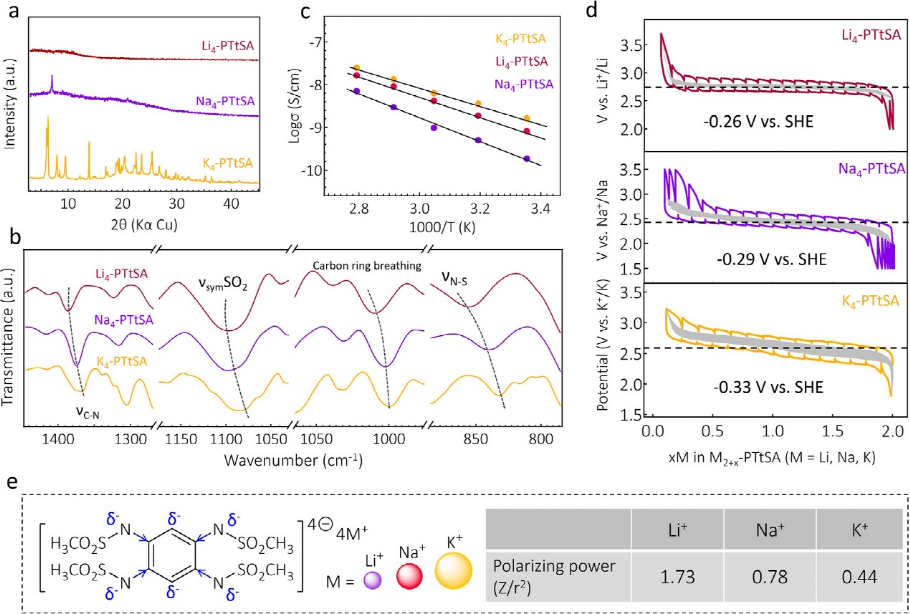
�D 2. Li4-PTtSA��Na4-PTtSA��K4-PTtSA�ı��^������(a) PXRD�Y�����о���(b) �c늺��ܶȺ�����߀ԭ�λ���P���ض����ε�FTIR��(c) 늌��ʵ�Arrhenius�D��(d) M4-PTtSA(M = Li��Na��K)늳���GITT�������ð�늳��M�Мy������늘O�������Ľ��١��Ӱ�^���ʾ�ӽ�ƽ�������߀ԭ�λ��(e) ���в�ͬ��x�ӺͲ���늺ɵ�����x��PTtSA�Ǽܣ��{ɫ�����{ɫ���^��ʾ���w�T��Ч����+I�����҈D��ʾ�A������x�ӵĘO���������
���ڎ�����늺ɵ��x�Ӱ돽���ӣ��O�����ʣ�һ����늺��c�x�Ӱ돽֮�ȳɱ�����ֵ����Li+��Na+�ٵ�K+��u�pС��������о��ѽ����������^����x�ӵĘO�����������{������ܶȣ��Ķ��{������߀ԭ�λ������Ҳͨ�^��Li4-��Na4-��K4-PTtSA�ķ����C����ͨ�^�^������x���{������߀ԭ�λ���ձ��m���ԡ�
�D2b�@ʾ��νC-N��νsymSO2��̼�h������νN-S�����{�ƣ������@Щ�F����Զ�ͨ�^�A������x�ӵ��o�Ч������������߀ԭ���ĵ�����ܶȁ���ጡ��S���u�����@Щ���늌��ʣ��D2c�����M��늌��ʌ�늳ز������P��Ҫ�����ЙC늘O���ϵ�늌�����Ȼ���ٱ��о���Li4-��Na4-��K4-PTtSA��ı���늌������Ҝ��¾��_��10–8 S/cm�������@Щ���ϵķ��ӽY�����A�ڵ��ǘӣ�늌����c�ضȴ�����ه�Pϵ���@��ӳ��һ�N�댧�w��͵ğἤ���ݔ�����@Щ�A�����ЙC��������߀ԭ�錧��늺ɂ�ݔ���Ա��ų��������S�Ϳ������������늺ɂ����C�ơ���Ȥ���ǣ�Li-Na-K�@һЩ�����늌��ʛ]�����@��څ�ݣ��@�����Ǹ��ƵĽY���Ⱥ��a��늺��x/���ķ��ӶѯB֮�g�ď��s�������ɵġ����ú�����gЪ�ζ����g(GITT)�y��Li4-��Na4-��K4-PTtSA�������߀ԭ�λֵ����ͨ�^���^����x���u������ܶ�׃����Ӱ푡��P������߀ԭ�λ�����y�����cLi+/Li0��Na+/Na0��K+/K0���Pϵ��D2d��ʾ�����˸��õı��^��ƽ����Ӌ��Li4-��Na4-��K4-PTtSA������߀ԭ늄��c�˜ʚ�늘OSHE��ȣ��քe��-0.26��-0.29��-0.33 V��
��D2d�еĔ�����ʾ��������PTtSA����߀ԭ���ĵ�����ܶȕr������߀ԭ�λ�����@���½�څ�ݣ��@���������A������x�ӵĘO��������������ġ��mȻ�o���@�þ��_�ľ��w�Y���������Ժ����ؼ��O����ȫ�����������ʣ��ăɂ��A������x�ӌ��ЙC����߀ԭ��Ԫ�������õ��o�Ч�������҃ɂ����Ƅӵ���x�Ӿ��п�׃�ġ��ɷ���ه��Ч����
���˸��õ��˽ⲻͬ�A������x�ӌ�����߀ԭ�λ��Ӱ푣�߀�M����DFTӋ�㡣�Y��������M4-PTtSA(M = Li��Na��K)�в�ͬ������߀ԭ�λ�cπ�h�����|�Լ�Li+��Na+��K+���ؓ�Բ���P���A������x���cPtTSA���ӵ���λ�������ĭh�IJ�ͬ�̶ȵ�ȥ�������ͷ����Ը��ԡ��D2e�o����ʾ��D�����ЉA������x�ӵIJ�ͬ�O��Ч���ṩ�˲�ͬ��ʩ���Б�Ч��(+I)��
���⣬M4-PTtSA(M = Li��Na��K)�@ʾ���߲��������ʣ�> 90�����͵͘O����<200 mV�������������W���٣��@���ܲ��֚w�����������еČ�����Լ��ЙC��܃ȸ�������x�ӔUɢ���D2c)�����w���ԣ����^�Y�����H��ʾ��M4-PTtSA����A�x��늳����O���ϵ��ձ��m���ԣ����Ҟ�ͨ�^���^����x�ӽ��Q�{���ЙC늘O���ϵ�����߀ԭ�λ�ṩ����Ҫָ����
3. ��늳�����
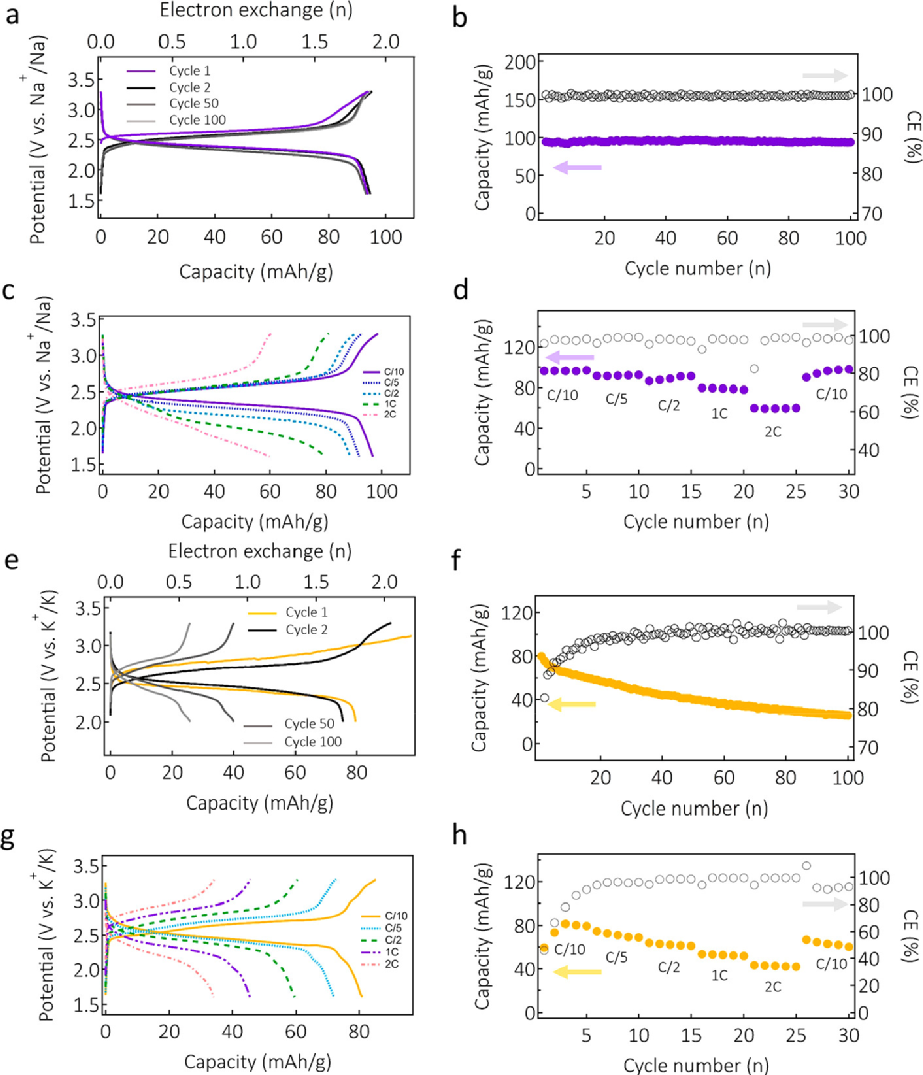
�D 3. Na��K��늳���늘O���ϵ�늻��W���ܡ�(a)��C/10�r�x��ѭ�h��Na4-PTtSA/Na늳غ��������������(b) ����늳ص��L��ѭ�h���ܡ�(c) ���������� (d) Na4-PTtSA/Na늳���C/10��2 C�����ȵĸ��N����ܶ��µı�������ѭ�h������(e)��C/10�r�x��ѭ�h��K4-PTtSA/K늳غ��������������(f) ����늳ص��L��ѭ�h���ܡ�(g) ���/���������(h) K4-PTtSA/K늳ر������ܵ�ѭ�h����������ܶȷ�����C/10��2 C����
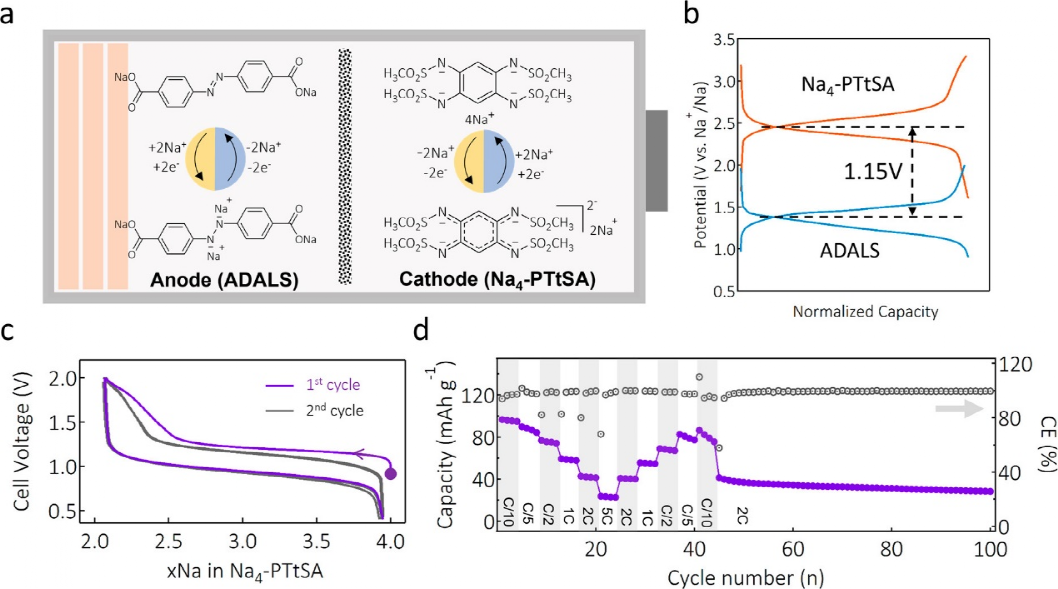
Na4-PTtSA/Na늳���1.7��3.3 V(vs Na+/Na)���λ�������M�еĺ����ѭ�h�yԇ������ܶȞ���5С�r�Ƚ��Q1��Na+���ĈD3a���Կ�����늳���2.5 V vs Na+/Na̎�@ʾ��ƽ̹�ij��/���ƽ�_�������������95 mAh g–1��������ÿ������ʽ��Ԫ���Q�˴�s1.9������Na��ֵ��ע����ǣ�2.5 V vs Na+/Na�������ֹ���c��������ֱ�Ӻϳɣ�synthesized in the Na-reservoir phase�����c�x���ЙC���O�����е����ֵ����ʹ������n���ЙC�������У���ֻ֪������-1,4-��������Na+/Na����2.72 V�ĸ����λ����������늽��|�еĸ��ܽ�ȣ��@�N������������M�з�����ѭ�h�����L��ѭ�h�У�Na4-PTtSA/Na늳ر��F���@����ѭ�h�����ԣ� 100��ѭ�h�����������ʞ�99.7%��ƽ�����Ч��≥99.5%����������֪���@�������ֹ���������SIB���ЙC���O�����ѭ�h�������D3b����
K4-PTtSA�������O����Ҳ��K4-PTtSA/K늳����M�����u��������ٴΏ��{���ǣ�K4-PTtSA�������ֹ����ĵ�һ�NK�����ЙC���O���ϣ�K-reservoir organic cathode�����D3e�@ʾ��K4-PTtSA/K늳ص�늉�-�λ��������5С�r�Ƚ��Q1��K+�ı���ѭ�h���cLi4-PTtSA��Na4-PTtSA늘Oֻ��һ��ƽ�_��ȣ�K4-PTtSA�@ʾ���ɂ�����߀ԭƽ�_��ƽ����2.6 V vs K+/K���D3e����늳��������_��80 mAh g–1������ÿ������ʽ��Ԫ�M����1.8��K+�Ŀ��潻�Q����C/10��ѭ�h100�κ�ѭ�h���^�ɂ��£��������˽ӽ�50%�ij�ʼ�������D3f����ֵ��ע����ǣ����Ч����ǰ�ׂ�ѭ�h���g�dz��ͣ���50��ѭ�h�����ӵ�100%��ͨ����늘O�ܽ����ЙC늘O��������˥�p����Ҫԭ��֮һ��������K4-PTtSA늘O���t�ų����@��ԭ�����ԓ��������ѭ�h�^���и������ܽ⡣늳���1 C�� 2 C�ĸ�����ܶ��¿ɾS��45��35 mAh g–1��~50%��40%����Փ������������������ K+���^��ߴ���^���ĔUɢ��չ�F���������Ե���Na+늳ء�
4. ԭλXRD����
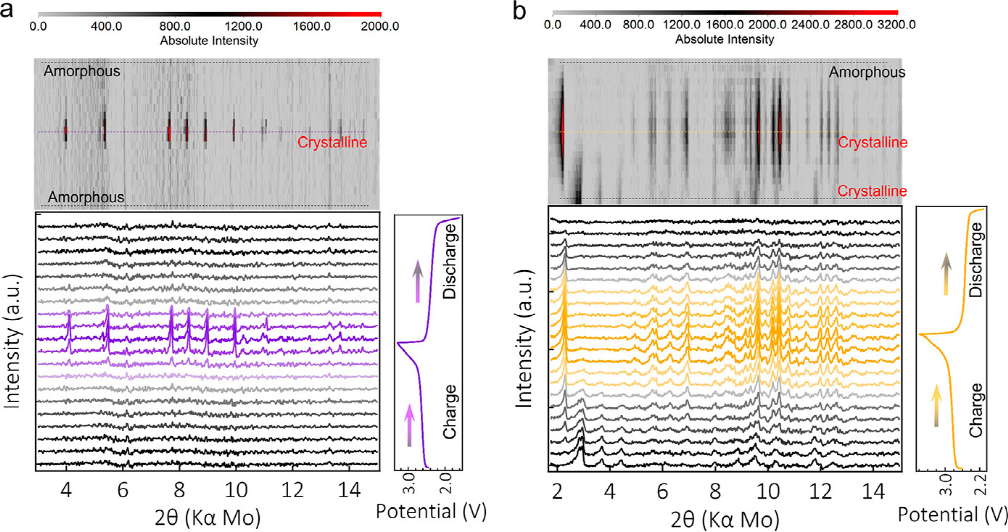
�D 4. (a) Na4-PTtSA �� (b) K4-PTtSA늘O���״γ��/���ѭ�h��ԭλXRD�����S�ȸ߾��D�@ʾ�ڈD��픲����ײ��@ʾ�˲�ͬ���늠�B�µ�ԭλXRD���D���҂��@ʾ�������ĺ��������������
����ǰ���ڈD2a����ӑՓ�ģ�Na4-PTtSA�ʬF���^��ĽY���Ի�Ǿ��࣬�@Ҳ��ԭλXRD�y�����^�쵽���D4a�������״γ���_ʼ�r��Na4-PTtSA���֟o���Σ����ڽӽ��״γ�늽Y���r�^�쵽�Y����ij��F��������Na2-PTtSA������4.1°��5.5°��7.7°��8.3°��9.0° 10.0°̎���F�ˎׂ��塣���Mһ��ѭ�h�Y�����ڷ�늵������A����u��ʧ����������ȫ�c����Ǿ�����Ա�������Ȥ���ǣ��M��Na4-PTtSA���Пo�������|����ԓ������ĽY����늻��W����ġ�����K4-PTtSA늘O��ԭʼ�������Y���࣬��2.8°��2.9°��3.7°��4.4°��̎�����壬�@��ԭλ�y���ĵ�һ��PXRD�D�п��^�쵽���D4b�����ڳ���^���У�ԭʼ����u��ʧ����ͨ�^�p����w���������õĽY������K2-PTtSA��ȡ����Ȼ�����S���Mһ����늣�ԓ�Y�������֏͵�ԭʼ�Y������ǷǾ���K4-PTtSA�ࡣ��M4-PTtSA��M = Li��Na��K���У���ȫ����������BM2-PTtSA��M = Li��Na��K���ǽY���ģ�����ȫ߀ԭ������늻��W�����^����ͨ���ǟo���εģ��@�����������~��ĉA������x�ӵ�λ�e��ɵĽY��׃�Ρ�
5. ȫ�ЙC늳�����
�D 5. (a) ȫ늳ؽY��ʾ��D��ؓ�O�����O�е�����߀ԭ�C�ơ�(b) Na4-PTtSA��ADALS�ĺ���������������ڰ�늳��cNa���ق΅���늘O�͌�늘O�Мy����(c) NAOSIB��0.1 C�µ�ǰ�ɂ�ѭ�h늉�������(d) NAOSIB ԭ��늳��ڲ�ͬ������ѭ�h�ı������cѭ�h�Δ���
������CM4-PTtSA����A�����x�����O���m���ԣ������Ƃ��˻��ڵ�����߀ԭ��ȫ�ЙC�c�x��늳�(NAOSIB)���D5aչʾ��NAOSIBԭ��늳صĽY�����\��ʾ��D������Na4-PTtSA�������O���ϣ�ż����-4,4'-��������}(ADALS)����ؓ�O���ϣ��D5b����ż�����F�Ŀ�������߀ԭ���S��1.3 V vs Na+/Na��ƽ���λ�惦�ɂ�Na+����ˣ���ؓ늘O���Ͼ����S�ɂ�Na+����ش�����D5c��ʾ��NAOSIB��ƽ��ݔ��늉��s��1.15 V��늉��������ЃAб��NAOSIB������C/10��ѭ�h�������ṩ1.9����Ӯ����Ľ��Q�������s95 mAh g–1���������O���ϵ��|�����D5c�������NAOSIB�@ʾ�������Ĺ������ܣ��@�ڏ�C/10��5 C�IJ�ͬ�����¾��з�����ѭ�h�����õ��������������C�����D5d������ʹ��1 C�ĸ�����ܶ���Ҳ���и���60 mAh g–1�����������ʡ������c�x��ȫ늳��⣬K�x��ȫ늳�Ҳ����ͨ�^��K4-PTtSA���O�����c��ͬ��͵��ЙCؓ�O������ADALS�������������⛵ȣ��M��ƥ�䡣
���YՓ��
��֮������x��PTtSA��ܱ��C����������ͨ�õ��ЙC���O�����ڸ����܉A�x��늳أ������ֹ����䇡��c��⛣����@�N���W�������H��SIBs��PIBs�I��dz��·f��߀�ǵ�һ�����_���c�x�Ӻ���x�Ӄ����ЙC���O���ϡ�Na4-��K4-PTtSA����2.5 V vs Na+/Na��2.6 V vs K+/K������߀ԭ�λ�£��քe�@ʾ���ɂ���Na+��K+�Ŀ��潻�Q���Ҿ��Ѓ�����ѭ�h�����Ժ������ܡ���Li4-��Na4-��K4-PTtSA���ϵ�y�о��У����ߴ_���ˌ�����߀ԭ������ֱ��Ӱ푵ĽY������늺ɶ�λڅ�ݡ��ЙC늳ز��ϵĸ߱���늌��ʎ��]�б��о��^�����܉F���ٵĄ����W����ֵIJ������ú͵�늘O�O�����@Щ�����ڌ��H늳ز��ϵ������Ђ��������@헹����нM�b�˵�һ����ڵ�����߀ԭ��ȫ�ЙC�c�x��늳�ԭ�ͣ�ݔ��늉���1.15 V���������ܸ��_5 C���@ʾ����ɫ��ѭ�h�����ԡ��mȻ�@һ���O���Ͽ����ԟo���c�o�C���ϸ���������Ӡ���������������ʿ�{���O��Ȼ�����c���^��ʮ��ďV���о����_�l�̓����ğo�C���O��ȣ���һ��PTtSA���O�ѽ��@ʾ�����õ�늻��Wǰ����߀�кܴ��̽�����M���g�����w���ԣ��@��о����H�S�������d�ĸ߉�SIB��PIB�ЙC���O���ώ죬߀�S����������һ�����ܵĉA�x�ӻ��W�wϵ��늳غ��O���֪�R���@헹�����A�x���ЙCꎘO�I���_�����µ�;����
Jiande Wang, Xuelian Liu, He Jia, Petru Apostol, Xiaolong Guo, Fabio Lucaccioni, Xiaozhe Zhang, Qi Zhu, Cristian Morari, Jean-François Gohy, and Alexandru Vlad*, A High-Voltage Organic Framework for High-Performance Na- and K-Ion Batteries, ACS Energy Lett. 2022, https://doi.org/10.1021/acsenergylett.1c02571




�����Wע�� ����Դ��XXX�����Ї�늳��ˣ�������Ʒ�����D�d������ý�w���D�dĿ�����ڂ��f������Ϣ�������������Wٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ���ݡ������������}��Ҫͬ���Wϵ�ģ�Ո��һ�܃��M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺[email protected]
-
���u��Դ��������Ԫ���Ԙ������������Ե��ЙC���늳�
2021-11-03 18:44 -
���Ҽ{���������ЙC̫���늳ؽ�������о�����ȡ�����Mչ
2021-10-12 18:40 -
���ЙCEV늳ز����Ҵ��܄�����������ܶ�
2021-06-22 10:01 -
��Nature���������ЙC���ɻ�늳�
2021-05-10 10:30 -
�ƌW���_�l�ЙC�o����늳� ��������Һ�н����ɻ�������
2021-05-08 10:28 -
�ձ�������˾PJP Eye�_�l̼늳� ���Ï��ЙC���л��յ�̼����
2021-04-14 10:54 -
�ЙC�������늳��а�����Щ��Ҫ��ɫ��
2020-11-30 11:33 -
����аl�ЙC����߀ԭҺ��늳� ������ɽo늄���܇���
2020-11-23 08:32 -
���������x��Һ�wȡ�����y�ЙC늽�Һ �аl����ȫ/���־Ã����O��
2020-09-08 08:58 -
ʥ���_��W��ˮ���}늽�Һ���늳ص��ЙC�܄� ����늳سɱ�
2020-06-02 19:15
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



-
���u��Դ��������Ԫ���Ԙ������������Ե��ЙC���늳�
2021-11-03 18:44 -
���Ҽ{���������ЙC̫���늳ؽ�������о�����ȡ�����Mչ
2021-10-12 18:40 -
���ЙCEV늳ز����Ҵ��܄�����������ܶ�
2021-06-22 10:01 -
��Nature���������ЙC���ɻ�늳�
2021-05-10 10:30 -
�ƌW���_�l�ЙC�o����늳� ��������Һ�н����ɻ�������
2021-05-08 10:28 -
�ձ�������˾PJP Eye�_�l̼늳� ���Ï��ЙC���л��յ�̼����
2021-04-14 10:54 -
�ЙC�������늳��а�����Щ��Ҫ��ɫ��
2020-11-30 11:33 -
����аl�ЙC����߀ԭҺ��늳� ������ɽo늄���܇���
2020-11-23 08:32
-
���ݹɷ��c�|����ܺϽ�늳ظ�Ĥ�Ŀ����Ŀ�䰸
2022-01-08 16:01 -
�ձ��о��ˆT�_�l䇿՚�늳� �����ܶ��_��500 Wh/kg
2022-01-15 15:49 -
���J���Ƴ����Ͱ�ȫ�P�I늳� ���F܇�v���܌��r����
2022-01-12 09:16 -
����|�ӽ��QĤ�Gɫ���ܘ˜�ӑՓ���ڷ�ɽ���_
2022-01-11 11:25 -
���ۄ���늳ػ��ա�һ��һ���r������ע����Iͬ����26���ИI���ٔU��
2022-01-10 10:42 -
�̑B늳���܇���������ǂ������ġ��e�ԡ�
2022-01-29 13:44 -
늳r�����2022���ϝq
2022-01-14 22:58 -
���r���ġ�ȫ��Ͱ��Ұ��
2022-02-04 14:16

�rֵ�ɾ��ИIƷ�ƣ����\�����ṩ���������YӍ
��ICP��09081210̖
 ��I��̖
��I��̖ �Ź���̖
�Ź���̖