

��(d��ng)ǰλ��: 늳�(li��n)�˾W(w��ng) > ǰ�� >
Chem. Mater.��Ҋ�⣺�늳�?z��)o����O����LMNO����(y��ng)�C(j��)��
�r(sh��)�g:2022-08-23 11:31��Դ:��Դ�W(xu��)�� ����:Energist
�c(di��n)��:
��

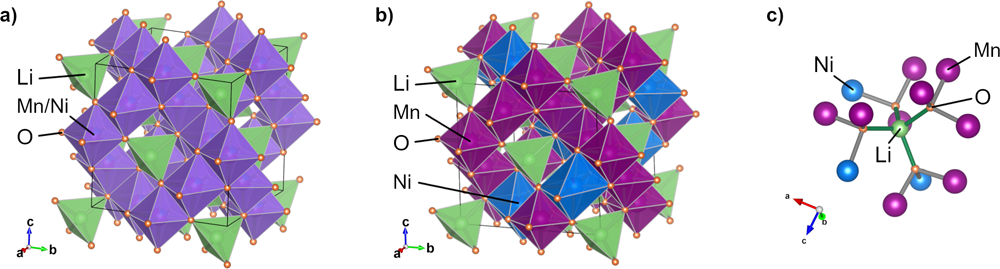
�D1.��a����Fd3̅m���gȺ�����е�TM�o��⾧ʯLNMO�ͣ�b��P4332���gȺ��TM����⾧ʯLNMO�ľ��w�Y(ji��)��(g��u)ģ�ͣ���c����������TM�⾧ʯLNMO�ľֲ�ԭ����λ�Y(ji��)��(g��u)��
LNMO-O��LNMO-D�ľֲ��Y(ji��)��(g��u)Ҳ��ͨ�^Raman���V�M(j��n)�Ѕ^(q��)�֣����D2a������493��637 cm−1����(sh��)�ķ���߹��У�λ��223��242��593 cm−1�������匦(du��)��(y��ng)������TM��ͬ�r(sh��)LNMO-O��~400 cm−1̎Ҳ���^��(qi��ng)��Eg�壻7Li�̑B(t��i)�˴Ź�����V�Á��о�LNMO-O��LNMO-D��Li�ľֲ��h(hu��n)�������D2b�У�LNMO-O��1010 ppm̎�ķ��ʾLi̎�چ�һ�h(hu��n)���У��~����F(xi��n)��935 ppm��834 ppm�匦(du��)��(y��ng)��Mn�ĭh(hu��n)���Լ�Mn3+��N������(du��)�ȣ�LNMO-D��600-1200 ppm���������^���ķֲ���935��834��736 ppm̎����(qi��ng)�ķ匦(du��)��(y��ng)Mn/Ni��������λ�����F(xi��n)��ȱNi�Ġ�B(t��i)����LNMO-O��Ʒ�в����ھֲ���Ni�ĭh(hu��n)����1088 ppm����
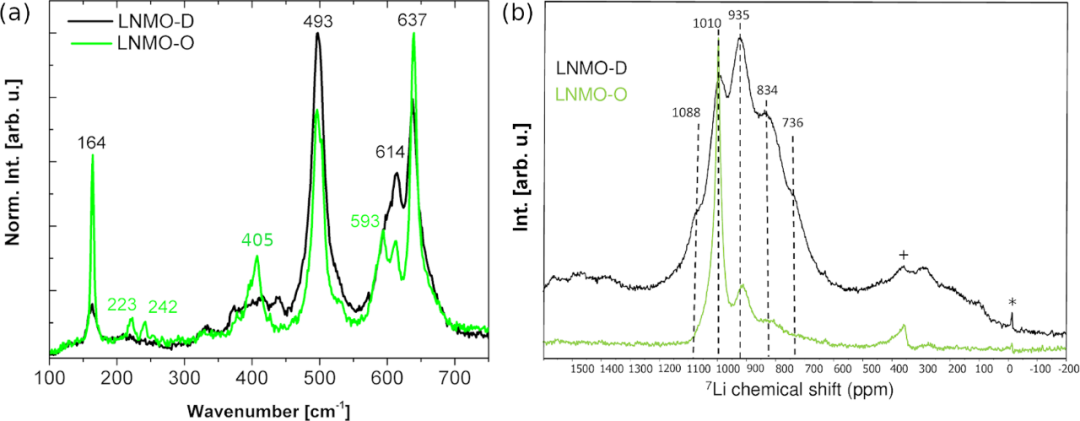
�D2.��a��LNMO-O��LNMO-D��Ʒ�Ěwһ��Raman���V�ͣ�b��7Li MAS NMR���V��
���˸����_�ر���LNMO-O��LNMO-D���ߵĽY(ji��)����B(t��i)�����߲������ӷ�ĩ���似�g(sh��)�M(j��n)���о����D3�����ɷN��Ʒ���������һ�µģ��ܺõ،�(du��)��(y��ng)�⾧ʯ�Y(ji��)��(g��u)����TM�������е�LNMO���p�ٵČ�(du��)�Q������(du��)��(y��ng)P4332���gȺ��Rietveld�����V�D�C��(sh��)LNMO-D�д��������Ďr�}�ͽY(ji��)��(g��u)����LNMO-O�ܺõ��cP4332�ļ⾧ʯ����(du��)��(y��ng)�����⣬Rietveld�������C��(sh��)�ɷN��Ʒ�ľ���(sh��)�s��8.18 Å���Դ����īI(xi��n)��(b��o)���Ĕ�(sh��)ֵ������Mn3+��N�Ĵ��ڌ�(d��o)�¾���U(ku��)��ͬ�r(sh��)LNMO-O��LNMO-D������100 nm���ҵĽY(ji��)���^(q��)��LNMO-D�ĸ�С��
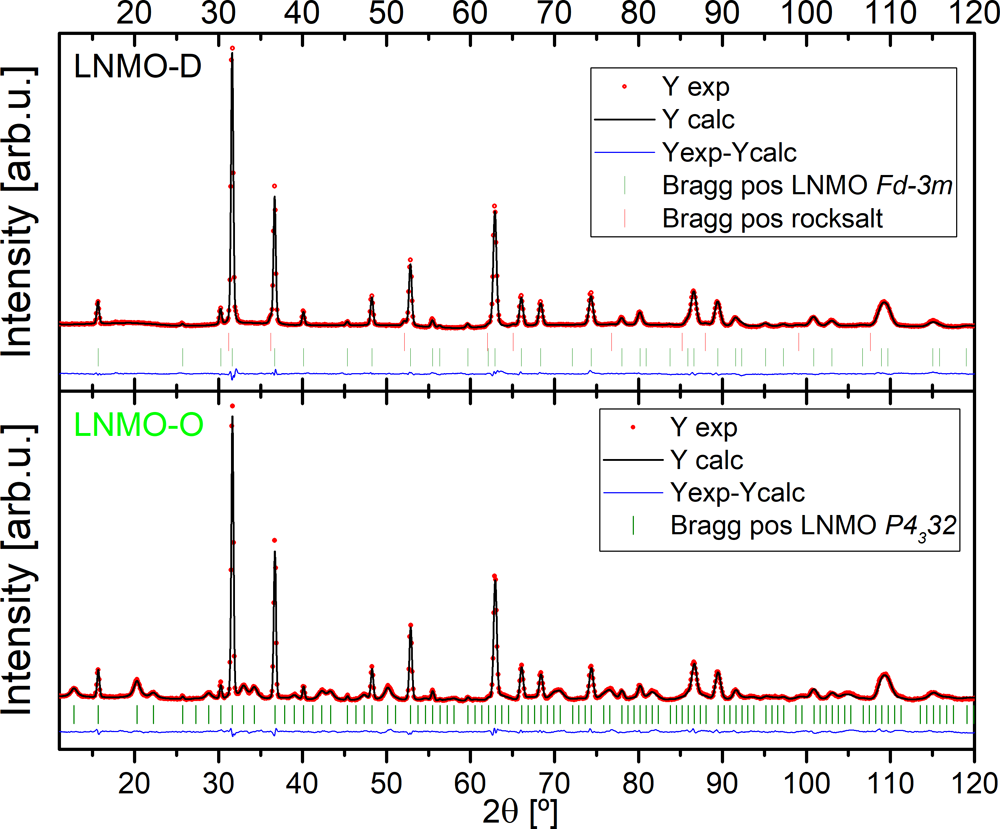
�D3. LNMO-D��LNMO-O��Rietveld���ވD�V��
2. Operando XAS����
����ֱ�^�ط���LNMO-D��LNMO-O��늻��W(xu��)�О飬���߲���Operando XAS���V�M(j��n)�б�����늻��W(xu��)�y(c��)ԇ늉���3.5-4.8 V��ѭ�h(hu��n)1.5Ȧ�����D4��ʾ��LNMO-D��4.0 V��4.8 V vs Li+/Li̎��늉�̎���F(xi��n)�ɂ�(g��)ƽ�_(t��i)��TM����K-edge��λ�ú͏�(qi��ng)��׃����ӳ��������(y��ng)�����Լ��ֲ��Y(ji��)��(g��u)��׃����Ni��#25̎�Ĺ��V�l(f��)��׃������(du��)��(y��ng)4.7 vs Li+/Li���λ����Mn��N��XAS���V��4.1 V̎�l(f��)��׃������(du��)��(y��ng)Mn3+/4+����߀ԭ��(du��)���״γ�늵�4.9 V�λ����(du��)��(y��ng)#105���V��׃������#180̎���һ��(g��)������ѭ�h(hu��n)���ڶ��γ����5V��ֹ늉��Դ_��TM����ȫ������ע�#290֮���XAS���Vֻ��С��׃������ʾ4.95 V��늉�ƽ�_(t��i)����Ҫ��(du��)��(y��ng)�����ĸ�����(y��ng)��
�����M(j��n)һ���Y(ji��)����Ҫ�ɷַ�����PCA���Ͷ�Ԫ�����ֱ���-������С���ˣ�MCR-ALS��������LNMO-D��XAS���V����ȡ��(sh��)��(j��)�M(j��n)�ж���������PCA�Y(ji��)������Ni K-edge��(sh��)��(j��)��(du��)��(y��ng)����(g��)��(d��)���ĽM�֣���Mn��(du��)��(y��ng)���ڃɷN�M�֣��D5�������_·늉������V<#4���£�����ԭʼ��һ��Ni��Mn�ĽM�֣���(d��ng)��늳��m(x��)���@Щԭʼ�M�ֺ�����u�p��ͬ�r(sh��)�M��2���ӣ���(du��)Mn���f���M��2��#105̎��4.9 V���_(d��)�����(du��)��(y��ng)�״γ�늽Kֹ?f��n)�B(t��i)��EOC1������Ni��N�ĽM��2���F(xi��n)��#67̎��ؕ�I(xi��n)60%�ij��������������һ��(g��)˲�r(sh��)����N����#67֮��M��2�D(zhu��n)׃?y��u)�M��3������#105̎�_(d��)����������@Щ�D(zhu��n)׃?c��)ڷ���^�����ǿ���ģ��ڵڶ��γ���^���У�Mn�ĽM��2��Ni�ĽM��3��#295̎�_(d��)�����(du��)��(y��ng)���γ�늽Kֹ?f��n)�B(t��i)��EOC2������#295֮��>4.95 V����K-edge���Vδ�l(f��)��׃�������m(x��)�ij�늷���(y��ng)���漰TM�������c������(y��ng)�l(f��)�����P(gu��n)��LNMO-O����N�����Sѭ�h(hu��n)�λ�������cLNMO-D��ƣ�������һ��(g��)�@���ą^(q��)�e������LNMO-O���״γ���^�����_(d��)����һ��(g��)���ߵĽ�ֹ늉���5 V����Ni XAS���V����EOD��B(t��i)��ȫ����M��3��
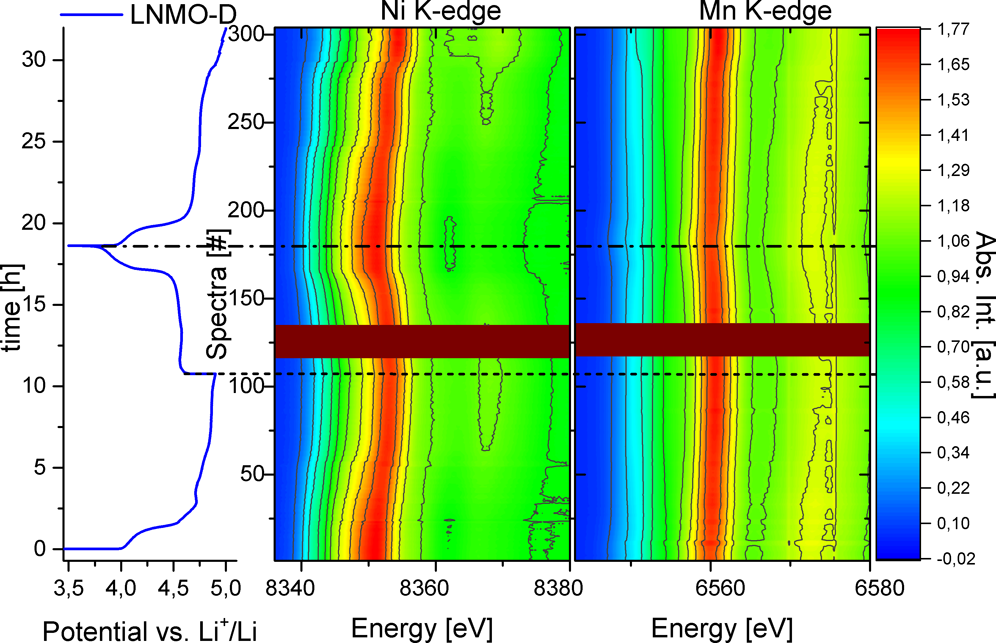
�D4. ��1.5��늻��W(xu��)ѭ�h(hu��n)��XAS��Ni��Mn K-edge����(du��)LNMO-D��Li��׃����
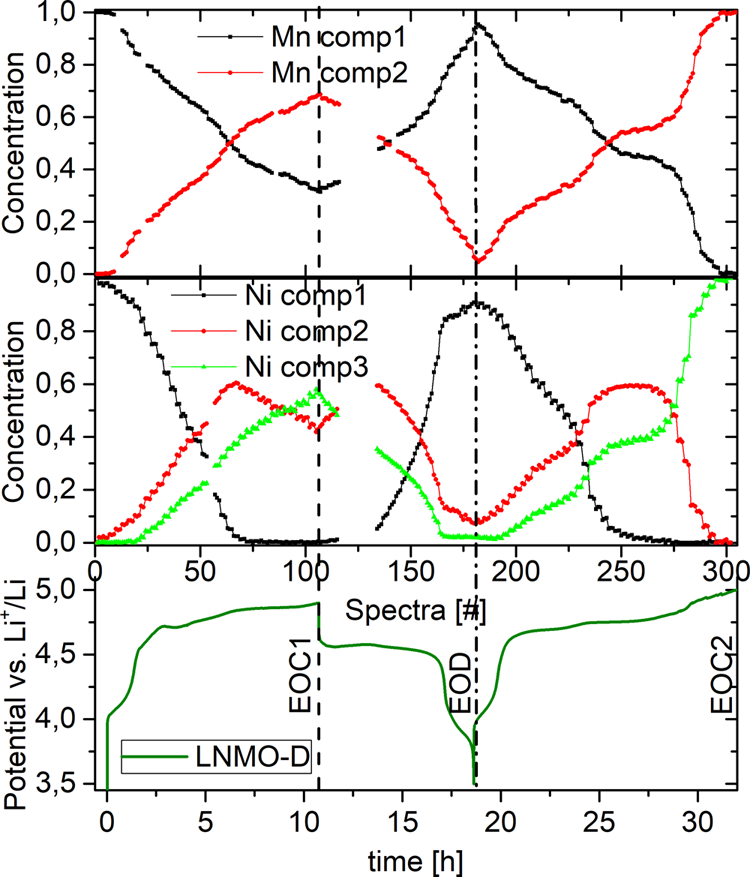
�D5. 1.5��ѭ�h(hu��n)LNMO-D��Mn��Ni�M�ֵĝ�ȷֲ���
3. LNMO�и��M�ֵ�XANES����
�D5�з����õ�LNMO-D��LNMO-O�и��M�ֵ�X�侀���ս�߅�Y(ji��)��(g��u)��XANES�����V���D6��ʾ�����ߵ�Ni�M��1��3�������Ƶģ�����Ni�M��2�����p��e��LNMO-O��Ni�M��2������߅����(du��)LNMO-D������0.2 eV���������ڸ����Ni3+�����B(t��i)������Mn��N������߅�c�������B(t��i)�������P(gu��n)�ģ���ˌ�(du��)20 eV̎��pre-edge�M(j��n)�з�����LNMO-D��Mn K-edge��pre-edge����d܉�����ѣ��Ķ��γ�6539.5 eV��t2g����6541.5 eV��eg���ɂ�(g��)�壬�ڳ���^���У����ڰl(f��)��Mn3+→Mn4+���D(zhu��n)׃��eg pre-edge�ķ及(qi��ng)���@�����ӣ���t2g��׃������(du��)��(y��ng)Mnλ�c(di��n)�ھ����е�Ť�������⣬��(du��)��LNMO-O���࣬��Mn K-edge pre-edge��LNMO-D����һ�£�����Mn������߀ԭ�����ڸ߶������TM������Ҳ���w�F(xi��n)��
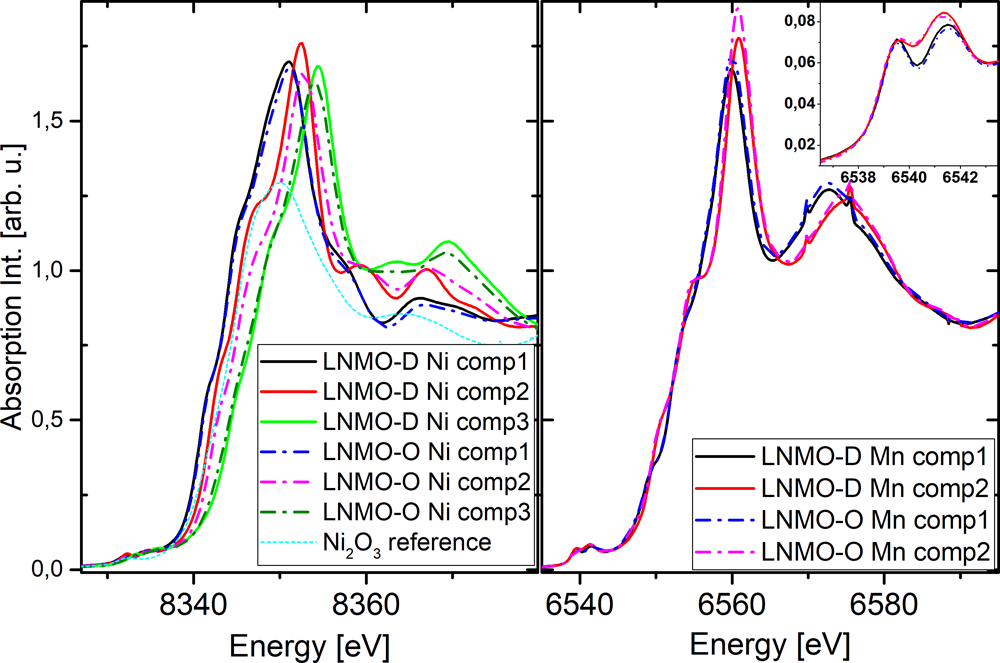
�D6. ��MCR-ALS��ȡ��ԭʼ���V��(sh��)��(j��)�������õ���XANES Ni��Mn K-edge�^(q��)��
���˫@��TM��Ԕ��(x��)�ľֲ��Y(ji��)��(g��u)׃���������M(j��n)���˔U(ku��)չX�侀���վ���(x��)�Y(ji��)��(g��u)��EXAFS���о�����(du��)MCR-ALS�Ы@ȡ�ĽM�ֺ��������M(j��n)�ДM�ϣ�����(g��u)���˳����^�̃�(n��i)��ԭ���cNi֮�g���x��ӛ��Ni-O1��Ni-TM���Լ�����(y��ng)��Debye-Waller���ӣ�σ2����׃���������D7�����ڳ���^���У�LNMO-D��Ni-O1��Ni-TM���g����u���ӣ��@������TM�������^�����x�Ӱ돽�s�̣�Mn-O6�����w��ƽ���g��δ�l(f��)���@��׃�����f��Mn3+→Mn4+���D(zhu��n)׃δӰ�Mn-O�I���L(zh��ng)�ȣ���σ2���ӵ�׃��Ҳ�ɵó�Ni-O6��#65̎���������(du��)��(y��ng)˲�B(t��i)Ni3+��N���γɣ�Ni-TM��#110̎��σ2���(du��)��(y��ng)EOC1��B(t��i)����LNMO-O��ƽ���I�Ͼ��x��LNMO-D�Զ̣��@�cP4332���С�ľ���(sh��)���P(gu��n)��
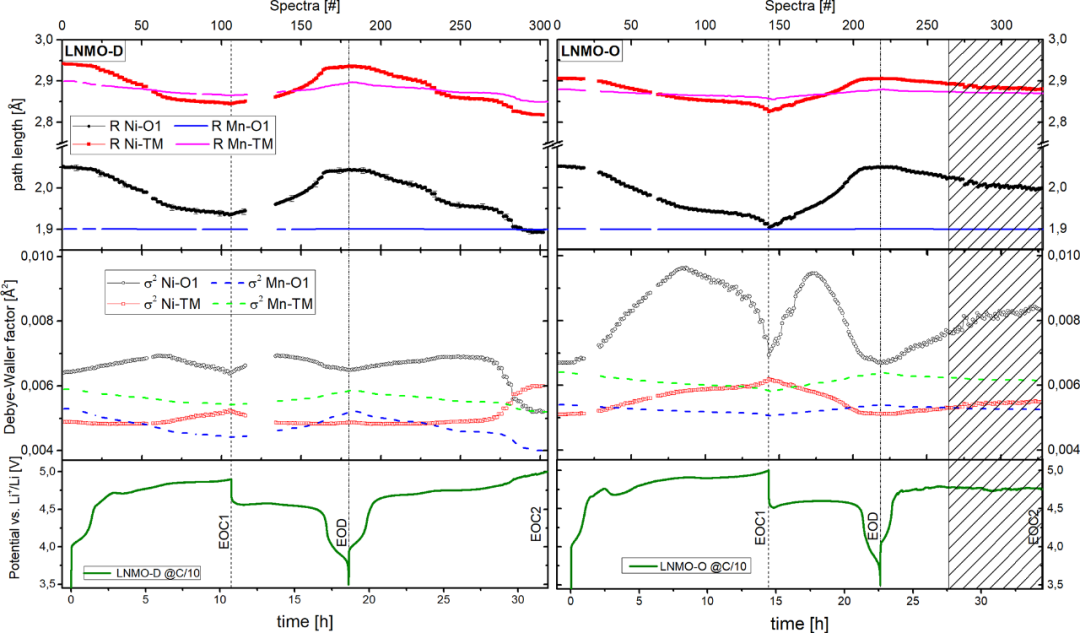
�D7. 늻��W(xu��)ѭ�h(hu��n)�^����LNMO-D��LNMO-O�������ɂ�(g��)Ni��Ni-O1��Ni-TM����·���L(zh��ng)�Ⱥ�Debye-Waller���ӵ�׃����
���Y(ji��)Փ��
���Ľ���Operando XAS��(du��)�ȷ�����TM����/�o�Č�(du��)Li��늻��W(xu��)����߀ԭ�C(j��)�ƣ��ɷN����LNMO-O��LNMO-D����ò�������Լ����W(xu��)Ӌ(j��)���ȷ��������Ƶģ�ÓǶ䇷���(y��ng)�^�̵IJ������TM�������C��(sh��)TM����/�o��LNMO�Ļ��Ծ�������Ni2+/4+����߀ԭ��(du��)����LNMO-O��Ƕ䇷���(y��ng)��LNMO-D�ĸ������⣬����ڟo��LNMO-D��˲�B(t��i)Ni3+��N���γ�����������LNMO-O�ğo���ԣ�����һ��Li�x��늳�LNMO�⾧ʯ���ϵ��_�l(f��)�ṩ��Ҋ�⡣
���īI(xi��n)��Ϣ��
Marcus Fehse,* Naiara Etxebarria, Laida Otaegui, Marta Cabello, Silvia Martín-Fuentes, Maria Angeles Cabañero, Iciar Monterrubio, Christian Fink Elkjær, Oscar Fabelo, Nahom Asres Enkubari, Juan Miguel López del Amo, Montse Casas-Cabanas, and Marine Reynaud, Influence of Transition-Metal Order on the Reaction Mechanism of LNMO Cathode Spinel: An Operando X‑ray Absorption Spectroscopy Study.
https://doi.org/10.1021/acs.chemmater.2c01360.
����(bi��o)����
�늳�
�o����O����




��؟(z��)�������ăH�������߂�(g��)���^�c(di��n)���c�Ї�(gu��)늳�(li��n)�˟o�P(gu��n)����ԭ��(chu��ng)���Լ�����������ֺ̓�(n��i)��δ��(j��ng)���W(w��ng)�C��(sh��)����(du��)�����Լ�����ȫ�����߲��փ�(n��i)�ݡ����ֵ��挍(sh��)�ԡ������ԡ����r(sh��)�Ա�վ�����κα��C����Z��Ո(q��ng)�x�߃H����������Ո(q��ng)���кˌ�(sh��)���P(gu��n)��(n��i)�ݡ�
�����W(w��ng)ע�� ����Դ��XXX�����Ї�(gu��)늳�(li��n)�ˣ�������Ʒ�����D(zhu��n)�d������ý�w���D(zhu��n)�dĿ�����ڂ��f������Ϣ�������������W(w��ng)ٝͬ���^�c(di��n)�͌�(du��)���挍(sh��)��ؓ(f��)؟(z��)��
������Ʒ��(n��i)�ݡ����(qu��n)���������}��Ҫͬ���W(w��ng)(li��n)ϵ�ģ�Ո(q��ng)?ji��n)�һ�܃?n��i)�M(j��n)�У��Ա��҂����r(sh��)̎����
QQ��503204601
�]�䣺[email protected]
�����W(w��ng)ע�� ����Դ��XXX�����Ї�(gu��)늳�(li��n)�ˣ�������Ʒ�����D(zhu��n)�d������ý�w���D(zhu��n)�dĿ�����ڂ��f������Ϣ�������������W(w��ng)ٝͬ���^�c(di��n)�͌�(du��)���挍(sh��)��ؓ(f��)؟(z��)��
������Ʒ��(n��i)�ݡ����(qu��n)���������}��Ҫͬ���W(w��ng)(li��n)ϵ�ģ�Ո(q��ng)?ji��n)�һ�܃?n��i)�M(j��n)�У��Ա��҂����r(sh��)̎����
QQ��503204601
�]�䣺[email protected]
����ϲ�g
-
����Science�ӿ��̑B(t��i)늳����ܺ��R������ܶȾ���(chu��ng)�o(j��)䛣�
2022-08-04 11:03 -
Jeffrey Moore�ȡ�JMCA�����x���ЙC(j��)Һ��늳�늽�Һ���Ͽɽ�������
2022-03-21 11:25 -
�㹤��&��������Science��䇽���늳،�(sh��)�F(xi��n)���L(zh��ng)������
2022-02-18 11:03 -
����䇽��ٮa(ch��n)Ʒ�����x��늳ص����ܡ���ȫ�ԺͿɳ��m(x��)��
2021-11-11 09:44 -
UNIST�_�l(f��)�oĤCO₂늳� �ɿ�����Ч�خa(ch��n)������
2021-03-05 11:21 -
GIST���ò���⒴����������늳؉���
2021-03-03 08:56 -
����&H.N. Alshareef���oؓ(f��)�Oˮϵ�\-�������i늳�
2021-01-22 11:37 -
���ϴ�W(xu��)��Angew. Chem. Int. Ed.��(b��o)��䇽���늳����M(j��n)չ
2021-01-06 11:45
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

���}


���P(gu��n)��
-
����Science�ӿ��̑B(t��i)늳����ܺ��R������ܶȾ���(chu��ng)�o(j��)䛣�
2022-08-04 11:03 -
Jeffrey Moore�ȡ�JMCA�����x���ЙC(j��)Һ��늳�늽�Һ���Ͽɽ�������
2022-03-21 11:25 -
�㹤��&��������Science��䇽���늳،�(sh��)�F(xi��n)���L(zh��ng)������
2022-02-18 11:03 -
����䇽��ٮa(ch��n)Ʒ�����x��늳ص����ܡ���ȫ�ԺͿɳ��m(x��)��
2021-11-11 09:44 -
Ampcera�����Ƴ�ȫ�̑B(t��i)늳ؼ��g(sh��) �Ɍ�(sh��)�F(xi��n)EV�����ٳ��
2021-04-16 08:40 -
UNIST�_�l(f��)�oĤCO₂늳� �ɿ�����Ч�خa(ch��n)������
2021-03-05 11:21 -
GIST���ò���⒴����������늳؉���
2021-03-03 08:56
�����c(di��n)
-
�ك|Ԫ��x��늳��(xi��ng)Ŀ�����������
2022-07-25 11:54 -
܇����늳أ���Ҫ�^���P(gu��n)
2022-07-24 10:15 -
�̑B(t��i)늳��ˣ����֛]��
2022-07-31 11:18 -
һ�K��(d��ng)��늳ص���������
2022-08-07 12:40 -
�䳲�̵���θҽа�ȁ��ϵ�Ƭ늳أ�
2022-08-08 08:40 -
��܇�����̵�����(j��)��늳�Ӌ(j��)��
2022-08-16 08:54 -
7�£��҇�(gu��)��(d��ng)��늳��b܇��24.2GWh��ͬ�����L(zh��ng)114.2%
2022-08-11 17:56
�gӭͶ��
(li��n)ϵ�ˣ���Ůʿ
Email��cbcu#www.astra-soft.com
�l(f��)���]���r(sh��)��@��Q#
�Ԓ��010-56284224
Email��cbcu#www.astra-soft.com
�l(f��)���]���r(sh��)��@��Q#
�Ԓ��010-56284224
�ھ�Ͷ��

©2017 ���(qu��n)���� 늳�(li��n)�� �A����̩�Ƽ�������������˾ ���k Power by DedeCms
�r(ji��)ֵ�ɾ��ИI(y��)Ʒ�ƣ����\(ch��ng)�����ṩ���������YӍ
��ICP��09081210̖(h��o)
�r(ji��)ֵ�ɾ��ИI(y��)Ʒ�ƣ����\(ch��ng)�����ṩ���������YӍ
��ICP��09081210̖(h��o)
 ��I(y��)��̖(h��o)
��I(y��)��̖(h��o) �Ź���̖(h��o)
�Ź���̖(h��o)