

�F�A���҇��Ј�����x��늳���Ԫ���i���O���ϴ��ڵĆ��}�c����
�r�g:2018-11-29 12:07��Դ:�²����ھ� ����:�C�ψ��
�c��:
��

�ڱ���ɹ��x��Ļ��W���������У���x��늳������乤��늉��ߡ�ѭ�h�����L�������ܶȸ��h���Ѻõ�һϵ��ͻ�����c�ɞ�ȫ���о����c����1991 �����ṫ˾�ɹ�����x��늳����F�̘I���ԁ�������Ӌ��C��ͨӍ�����M���ӮaƷ�I��đ���ȡ�ØO��ɹ���
Ŀǰ��x��늳ص���Ҫ�lչ�����������߹��ʣ�ͬ�r�@Ҳ����ӮaƷ�K�˲��������Q���Լ�����Դ늄���܇�ИI�lչ�Ѵ�ı�ȻҪ��
��ˣ�Ψ���^�m���ƬF����x��늳����P���g���_�l������x��늳ز��ϣ������m���������L�c�h�����o���f�{�İlչ������
�D1 ��x��늳�ԭ��D
1 ��Ԫ���O��������
��������O������߂��������ԡ�
��1�����ھ����^������߀ԭ�λ���װl������߀ԭ�������^�ɽ����x�ӣ��Ա��C��x��늳��^�ߵij���������ݔ��늉� ��
��2���^�ߵ���Ӽ���x��늌����Ա��C���õı������� ��
��3�����õĽY�������� ��
��4�����^��늉������Ⱦ����^�ߵĻ��W�����Ժ᷀͟���� ��
��5���^���Ƃ䣬�h���Ѻ��҃r���m�С�
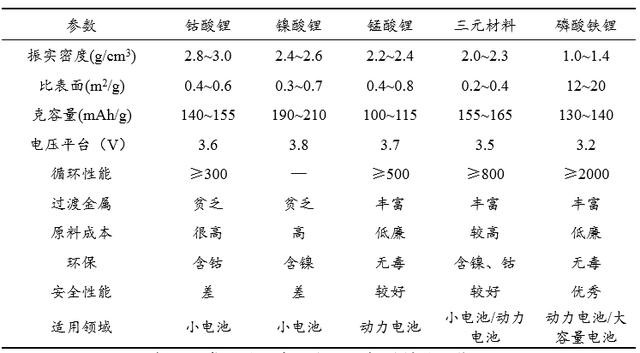
�D2 ������x��늳����O���ϵ����ܱ��^
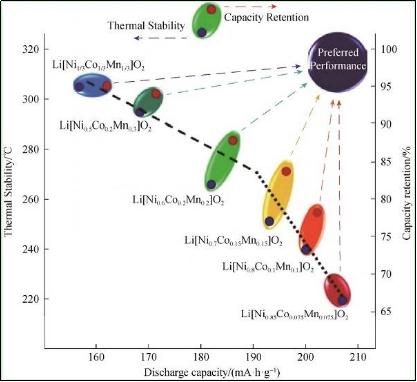
�D3 Li[Ni x Co y Mn z ]O 2 (NCM ,x =1/3, 0.5, 0.6, 0.7, 0.8, 0.85)�ķ���������᷀���Ժ������������Pϵ�D
��Ԫ���O����LiNi1-x-yCoxMnyO2��NCM�����б������ߡ��Y�������Ժá��᷀���Ժúͳɱ��^�͵����ԡ�LiNi1-x-yCoxMnyO2���Ͽ��J����Co�ӱ�Ni��Mn����ȡ��������Ni���������܉���߲���������������ѭ�h���ܺͷ����ԣ�Co������������������׃ ����߱������ܣ�Mn����������������߽Y�������ԣ������������������N�^�ɽ��ٵĺ����Q���˲��ϵĸ�����ܣ��״η�����������������ʡ��������᷀͟���Ե����ܟo��ͬ�r�_�������Ҫȡ�ᣬ��I�����a�^���п����m���Ľ���Ni���������Co��Mn ��ռ�ȣ�������ѭ�h���ܣ����L�aƷ������
1999�꣬Liu���״κϳɲ�ͬ�M�ֵ���Ԫ�^�ɽ������������LiNi1-x-yCoxMnyO2�������侧�w�Y����늻��W�����M��Ԕ���о����l�F��Ԫ����ѭ�h���������@���چ�һ�M�ּ���Ԫ�Ӡ���ϡ�2001�꣬Ohzuku�Ⱥϳɳ�Ni��Co��Mn ���NԪ�ص�����LiNi1/3Co1/3Mn1/3O2���ϣ���2.5V~4.6V늉������·�늱��������_200mAh g-1���˺����в�ͬ���i��������Ԫ���ϱ��_�l�ϳɡ���Ŀǰ��ֹ����Ԫ���O���ϵ��о����c��Ҫ�Ѓ�� ���� Ni��Mn �����ͣ���LiNi1/3Co1/3Mn1/3O2��NCM111����LiNi0.4Co0.2Mn0.4O2��NCM424�����ڸ���ͣ�����LiNi0.5Co0.2Mn0.3O2��NCM523����LiNi0.6Co0.2Mn0.2O2��NCM622����LiNi0.8Co0.1Mn0.1O2��NCM811����
�D4 LiNi0.6Co0.2Mn0.2O2��NCM622���Y��ʾ��D
2���چ��}�c����
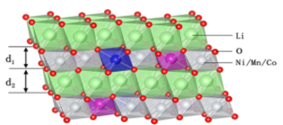
��Ԫ���O����NCM ����α-NaFeO2 �Ӡ�Y�������g�cȺ��Rm���ԭ�Ӻ��^�ɽ���ԭ�ӽ���ռ����ԭ�Ә��ɵİ����w����λ��NCM ��Rm �Ӡ�Y��������-䇌�-����-�^�ɽ��ٌ���б�������w[001] ����ѯB���ɡ��������w����Փ������e܉���ϵĹ������������Ni3+����������� Ni څ������Ni2+ ��ʽ���ڡ�����Ԫ�����Ƃ��^���У�����Ni2+�돽��0.069 nm���cLi+�돽��0.076 nm���ӽ��������ھ���Y���ИO�װl������ռλ���@�r䇌��c�^�ɽ��ٌ��g�������x�ӻ��ŬF��

�D5 α-NaFeO2 �����i��Ԫ���ϵĽY���D
���^������ČӠ�Y������x�ӻ��Ō��¾��w�Y����䇌��g���pС����x���w�ƻ�����ӣ�ͬ�rռ��䇌�λ�õ��^�ɽ����x��Ҳ��K����x�ӔUɢ����ˣ��S����x�ӻ��ŵ����ӣ����ϵı�������Ҳ�S֮������
���ϣ���Ԫ���O���ϵ���x�ӻ��ŬF�H���F����ϳ��^���У�������늳س���ѭ�h�rҲ���l����ͨ�^����R����ָ��O3 ���Ӡ����LixNi0.5Mn0.5O2ѭ�h�^���д����^�ɽ���Ԫ���w��ռ��䇌�λ�õ���r�����⣬�L���ڸ�늉�����ѭ�h��r��LiNi0.5Co0.2Mn0.3O2�l����׃����Rm ����u�D׃��⾧ʯ��͎r�}�ࡣ�ڸ߳�늽�ֹ늉��²������Ó䇣�����䇿�λ�Ĵ��ڌ��²��ϽY���O���������^�ɽ����x�ӏ��^�ɽ��ٌ��w��ռ��䇌�λ�ã���������ͬ�x��֮�g���ų�����ʹ�ðl�����ŵ���x�ӃA����ռ������䇿�λ��
� NCM ���O���Ͼ��и��������ͳɱ���ԭ�ρ�Դ�S���ȃ��c����һ�N�O�Б���ǰ������x��늳ز��ϣ�����ȡ�� LiCoO2 ����Ҏģ�̘I�������ǣ���懲��ϴ惦�^���������c���h���е�CO2��H2O �����M���ڲ��ϱ������ɺ�䇻���������늑B�¸��������Ե��^�ɽ����x�Ӵ�늽�Һ�ķֽ⣬����늽�Һ���ĵ�ͬ�r�ڲ��ϱ��������^��Ĺ̑B늽��|����ӡ����˵õ�����ČӠ�Y�����ϣ����������䇟��Y�Ƃ��^���м����^���Դ������NCM ���ϱ������䇚��������ϱ�����ڵ�䇚����c�՚���H2O��CO2��������Li2CO3��LiOH����懲����ڼ�ˮ�н���һ�Εr�g����ҺpH ֵ������12�����������NMP �܄��ИO���γ����z��{�ϣ�����Ӱ�늘O�OƬ������
���rLiNi0.7Co0.15Mn0.15O2�������s��150 nm ��һ���w���M�ɣ������❍�����ڿ՚��з���3���º�������Ҋ��䇻�����z�y�@ʾ�՚��д惦�����Li2CO3������0.89 wt%������1.82 wt%��LiOH������0.25 wt%������0.44 wt%�������Ԫ���ϴ惦�h����횴_�����^�՚��ˮ�֡���Ԫ�����ڳ���ѭ�h�^���л������|�c늽�Һ֮�g�����l�Ľ��渱������늘O���ϱ��渽������늽�Һ�ֽ����|���@Щ���|�ľ��w�M���c늳��wϵ��ʹ�õ�늽�Һ���P����������LiClO4����}��PC���܄��M�ɵ�늽�Һ�У� ��Ҫ�ֽ�a���̼��䇡�������LiPF6����}��EC��DMC���܄��M�ɵ�늽�Һ�У��ֽ�a��t��Ҫ�麬P��O ��F �Ļ�����ڲ�ͬ�yԇ�ضȡ��yԇ�r�g�Լ���늑B�£����O���ϱ����늽�Һ�ֽ�a����ɾ�̼������LiF��LixPFy ��LixPFyOz �M�ɡ����ϱ��渽����늽�Һ�ֽ�a����K��x���ڻ������|������w�ƣ������迹������늳�늻��W���ܐ�����ȥ������䇚����Լ����ƽ��渱�����Ǹ�����Ԫ����늻��W���ܵ��P�I��
3 �Y �Z
��1���F�Юa�I�������䇡������i��䇺������F��ڻ��A�о������ѽ��]�м��gͻ�ƣ��������ܶȺ��N��Ҫ���gָ���ѽ��ӽ��䑪�ØO�ޣ���Ԫ������δ���аl�ͮa�I���� �����������䑪���I��IJ�ͬ���քe����ܶȻ���늉����lչ��δ���İlչĿ���nj���Ԫ���ϵĉ����ܶ���ߵ� 3.9g/cm3���ϣ����늉��_��4.5 V������������_��200 mAh/g��늘O�����ܶȱ����䇸� 25%���Ķ�ȫ��ȡ�����䇣��ɞ�С��ͨӍ��С�̈́����I���õ��������O���ϡ�
��2���҇���С�̈́���늳��Ј�ǰ���V韣�������С�̈́���늳ص����a���_�l�T���^�ͣ��Դ����ϵ�y�^�麆�Σ�늳ذ�ȫ�������ơ���ˣ��YԴ�S�����r��������h���o���;Gɫ���ܵ���Ԫ���ό��ɞ���һ������늳ز��ϵ����x��
��3��ͨ�^���ƹ�ˇ�Ƃ����һ�Άξ��w������Ԫ�� ���܉�@�ø��������ľ��w�Y�����^�ߵĉ����ܶȺ̓�����늘O�ӹ����ܣ��ɞ�Ŀǰ��Ԫ��������S���ձ��о��İ�K��
��4����Ԫ���ϵ�ѭ�h�����c�����ஔ�������ܶȺͱ������܅s���������F䇣������������i��䇣����J������x�ӄ���늳����O���ϵ���Ҫ�x���ڄ���늳ؑ��÷��棬��Ԫ���ϳ��������������õľC������֮�⣬߀��ԓ������ͬ늳ص����������OӋ���a������ĮaƷ�����ڴ��̈́���늳أ����]�����ϰ�ȫ�ԣ��i���+��Ԫ���ϡ������F�+��Ԫ�����wϵ�^���m�ˣ�����С�̈́���늳أ��ɲ��ü���Ԫ�wϵ��
��5�����������ͬ���늳ص�ʹ�����c��ͨ�^�������iԪ�ر������Y�ϓ��s���������ԣ��_�l��������Ԫ���ϣ���������һ�����аl���c��
���ߺ���
�R�ɣ��������b��ɷ�����˾�U��x�ӄ���늳ػ����O���аl���̎������������������U�f��x�ӄ���늳ز��������x�ИI���꣬�߂��ۺ�Č��I���g�c�S�����ИIҊ�⣬������ڇ����������ڿ����s־���Wվ�l��늳ػ���̎�����P����Ӣ�����¡�




��؟�������ăH�������߂����^�c���c�Ї�늳��˟o�P����ԭ�����Լ�����������ֺ̓���δ�����W�C�����������Լ�����ȫ�����߲��փ��ݡ����ֵ��挍�ԡ������ԡ����r�Ա�վ�����κα��C����Z��Ո�x�߃H����������Ո���кˌ����P���ݡ�
�����Wע�� ����Դ��XXX�����Ї�늳��ˣ�������Ʒ�����D�d������ý�w���D�dĿ�����ڂ��f������Ϣ�������������Wٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ���ݡ������������}��Ҫͬ���Wϵ�ģ�Ո��һ�܃��M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺[email protected]
�����Wע�� ����Դ��XXX�����Ї�늳��ˣ�������Ʒ�����D�d������ý�w���D�dĿ�����ڂ��f������Ϣ�������������Wٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ���ݡ������������}��Ҫͬ���Wϵ�ģ�Ո��һ�܃��M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺[email protected]
����ϲ�g

���}


���P��
�����c
-
2024�늳����Ј��
2024-05-24 18:59 -
�������Ŀ�Kֹ���ذ��ļ�������I�μ��U��a�ܣ�
2024-05-15 19:12 -
С�����늳����죬�c���r���������Y��˾��
2024-05-20 19:05 -
�y�ֶ���������@����I5������늳��Ŀ�_��/���s!
2024-05-21 18:46 -
�ذ�������Դͻ���������ã��̑B늳�ِ���������l
2024-05-28 18:18 -
Ͷ�Y��25�|Ԫ���@������I�M���������O늳ػ��WƷ�Ŀ
2024-05-22 19:20 -
��һ10GWh�Ŀ�_�����̑B늳ؾ��x�a�I��߀Ҫ��ã�
2024-05-11 19:17 -
���r�����ȁ��ϡ��Є��º���ͬ��؛ε������������
2024-05-09 18:48

©2017 ������� �Gɫ�DžR��Դ���g�о�Ժ �A����̩�Ƽ�������������˾ ���k Power by DedeCms
�rֵ�ɾ��ИIƷ�ƣ����\�����ṩ���������YӍ
��ICP��2024061100̖
�rֵ�ɾ��ИIƷ�ƣ����\�����ṩ���������YӍ
��ICP��2024061100̖
 �Ź���̖
�Ź���̖